Key takeaways:
- The Lancet Commission identified high cholesterol and vision loss as new risk factors for dementia.
- The commission outlined 13 recommendations for individuals and governments to prevent dementia.
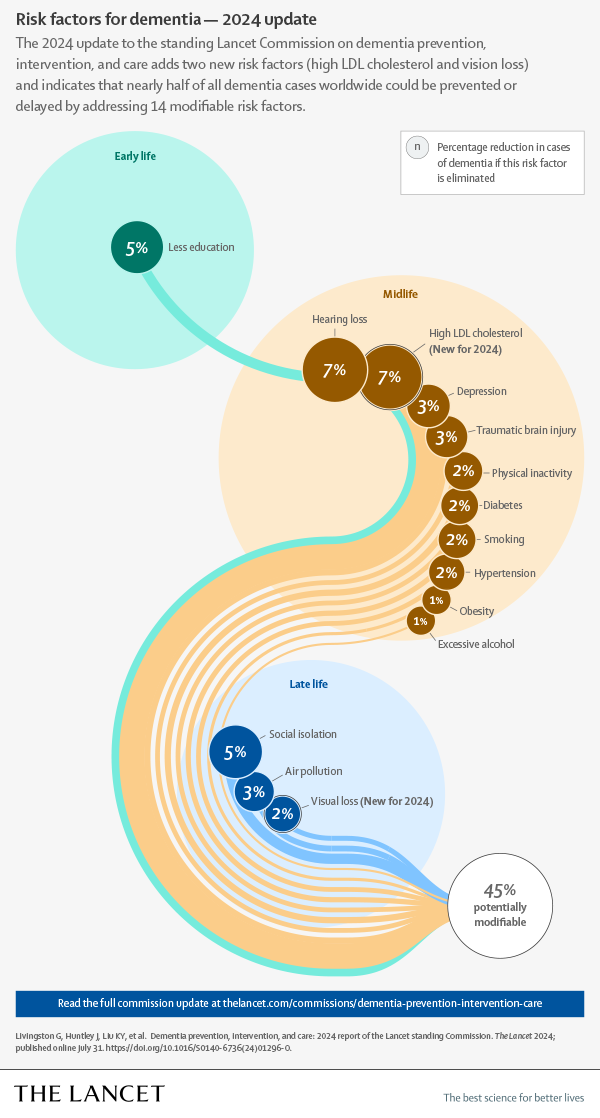
PHILADELPHIA — Tackling 14 risk factors for dementia beginning in childhood could prevent or delay nearly half of cases worldwide, according to a report from the Lancet Commission presented at the Alzheimer’s Association International Conference.
These include two risk factors — high cholesterol and vision loss — newly identified by the commission on dementia prevention, intervention and care.
An estimated 57 million people were living with dementia in 2019, Gill Livingston, MD, a professor of psychiatry at University College London, and colleagues wrote in the report. This number is expected to increase to 153 million by 2050, highlighting the need for risk reduction strategies.
The new report is an update to the commission’s 2020 report. Members of the commission adopted a triangulation framework that prioritized systematic reviews and meta-analyses. They also conducted new meta-analyses when necessary.
The researchers said their review supports the 12 potentially modifiable risk factors that were identified in the 2020 report: air pollution (RR = 1.1; 95% CI, 1.1-1.1); depression (RR = 2.2; 95% CI, 1.7-3); diabetes (RR = 1.7; 95% CI, 1.6-1.8); excessive alcohol use (RR = 1.2; 95% CI, 1-1.5); hearing loss (RR = 1.4; 95% CI, 1-1.9); hypertension (RR = 1.2; 95% CI, 1.1-1.4); lower education level (RR = 1.6; 95% CI, 1.3-2); obesity (RR = 1.3; 95% CI, 1-1.7); physical inactivity (RR = 1.2; 95% CI, 1.2-1.3); smoking (RR = 1.3; 95% CI, 1.2-1.4); social isolation (RR = 1.6; 95% CI, 1.3-1.8); and traumatic brain injury (RR = 1.7; 95% CI, 1.4-1.9).
The evidence also supports the addition of high LDL cholesterol (RR = 1.3; 95% CI, 1.3-1.4) and vision loss (RR = 1.5; 95% CI, 1.4-1.6).
If these 14 risk factors are eliminated, “nearly half of dementias could theoretically be prevented,” Livingston and colleagues wrote.
This has important implications for physicians, particularly family physicians, Livingston told Healio. She noted that diabetes, excessive alcohol use, hearing impairment, high LDL, hypertension, obesity, vision loss and smoking account for about one-quarter of all dementias.
“If we add depression, traumatic brain injury and physical inactivity, which family physicians also advise on, then it is a third of dementias,” she said. “Their active vigilance and advice potentially make a huge difference.”
Based on their findings, the researchers outlined 13 recommendations for individuals and governments to prevent dementia:
- ensure children have access to good-quality education and encourage individuals in midlife to participate in “cognitively stimulating activities;”
- reduce harmful noise exposure and make hearing aids accessible to those with hearing impairment;
- treat depression;
- promote helmets and other head protection during contact sports and when riding bicycles;
- encourage exercise;
- reduce smoking through education and by implementing policies that aim to control the cost of cigarettes;
- prevent or reduce high blood pressure;
- diagnose and treat high LDL;
- maintain a healthy weight and treat obesity early;
- reduce excessive alcohol use through price control and raising awareness about the risks of overconsumption;
- reduce social isolation by encouraging activities and living with other people, prioritizing an “age-friendly and supportive community, environments and housing”;
- ensure access to vision loss screening and treatment; and
- decrease air pollution exposure.
“Although addressing risk factors at an early stage of life is desirable, there is also benefit from tackling risk throughout life; it is never too early or too late to reduce dementia risk,” Livingston and colleagues wrote.
References:
- Livingston G, et al. Lancet standing commission on dementia prevention, intervention and care. Scientific advances in the 2024 commission. Presented at: Alzheimer’s Association International Conference; July 28-Aug. 1, 2024; Philadelphia.
- The Lancet: Nearly half of dementia cases could be prevented or delayed by tackling 14 risk factors starting in childhood, including two new risks — high cholesterol and vision loss. www.eurekalert.org/news-releases/1052982. Published July 31, 2024. Accessed July 31, 2024.
Perspective
Dementia risk reduction is an important area of research. In this latest Lancet Commission report, they’re adding two more risk factors — high cholesterol and vision loss — to the list and calculating that, together, these 14 factors could account for around half of all worldwide cases of dementia. This illustrates the importance of our awareness of these types of risk factors.
It’s important for our understanding that these reports are not just informed by epidemiological studies, but further interventional studies. One of those, which is ongoing, is the U.S. POINTER study. Recruiting is complete, but evaluating the results of the study is still underway. They will be reported next year at AAIC 2025 in Toronto. This study is looking at multiple risk factors, including modifying diet, exercise, cognitive activities, social engagement and the management of heart health status, and whether these factors in combination can protect cognitive health.
Of note, for right now, the Alzheimer’s Association provides 10 Healthy Habits for Your Brain, which is a great resource for anybody who is thinking about their risk.
Claire Sexton, DPhil
Senior director of scientific programs and outreach
Alzheimer’s Association
Disclosures: Sexton reports no relevant financial disclosures.
Source:
Livingston G, et al. Lancet. 2024;doi:10.1016/S0140-6736(24)01296-0.