by Jennifer Brown, University of Iowa
The genetic mutation that causes Huntington’s disease (HD)—a devastating brain disease that disrupts mobility and diminishes cognitive ability—may also enhance early brain development and play a role in promoting human intelligence.
This revelation comes from more than 10 years of brain imaging and brain function data, including motor, cognitive, and behavioral assessments, collected from a unique population—children and young adults who carry the gene for HD. While an HD mutation will eventually cause fatal brain disease in adulthood, the study finds that early in life, children with the HD mutation have bigger brains and higher IQ than children without the mutation.
“The finding suggests that early in life, the gene mutation is actually beneficial to brain development, but that early benefit later becomes a liability,” says Peg Nopoulos, MD, professor and head of psychiatry at the UI Carver College of Medicine, and senior author on the study published in The Annals of Neurology.
The finding may also have implications for developing effective treatments for HD. If the gene’s early action is beneficial, then simply aiming to knock out the gene might result in loss of the developmental benefit, too. Creating therapies that can disrupt the gene’s activity later in the patient’s lifetime might be more useful.
The new data about the gene’s positive effect on early brain development is also exciting to Nopoulos for another reason.
“We are very interested in the fact that this appears to be a gene that drives IQ,” she says. “No previous study has found any gene of significant effect on IQ, even though we know intelligence is heritable.”
HD gene linked to better brain development in early life
Huntington’s disease is caused by a mutation in the huntingtin (HTT) gene. The protein produced by the HTT gene is necessary for normal development, but variations within a segment of the protein have a profound effect on the brain.
The segment in question is a long repeat of one amino acid called glutamine. More repeats are associated with bigger, more complex brains. For example, species such as sea urchins or fish have no repeats, but these repeats start to appear higher up the evolutionary ladder. Rodents have a few repeats, while apes (our closest relatives) have even more repeats; and humans have the most.
Most people have repeats in the range of 10–26, but if a person has 40 or more repeats, then they develop HD. Although the gene expansion is present before birth, HD symptoms do not appear until middle age. Nopoulos’s team at the University of Iowa has a long history of studying how the HTT gene expansion affects brain development in the decades before disease onset.
“We know that the expanded gene causes a horrible degenerative disease later in life, but we also know it is a gene that is crucial for general development,” she says.
“We were surprised to find that it does have a positive effect on brain development early in life. Those who have the gene expansion have an enhanced brain with larger volumes of the cerebrum and higher IQ compared to those who don’t.”
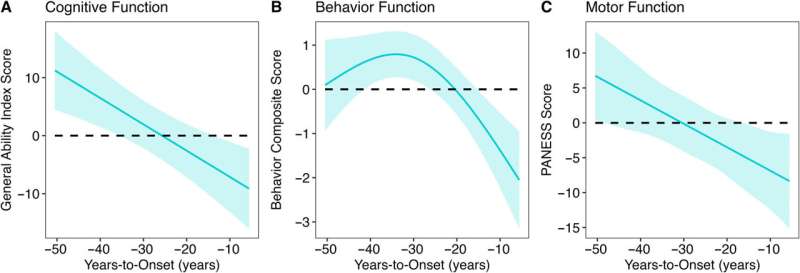
In particular, the study found that decades before HD symptoms appeared, children with the HD gene expansion showed significantly better cognitive, behavioral, and motor scores compared to children with repeats within the normal range. Children with the expanded gene also had larger cerebral volumes and greater cortical surface area and folding. After this initial peak, a prolonged deterioration was seen in both brain function and structure.
The study gathered this data by following almost 200 participants in the Kids-HD study, the only longitudinal study of children and young adults at risk for HD due to having a parent or grandparent with the disease.
Evolutionary benefit comes at a cost
Although surprising, the findings are in line with studies by evolutionary biologists who believe that genes like HTT may have been “positively selected” for human brain evolution. This theory, known as antagonistic pleiotropy, suggests that certain genes can produce a beneficial effect early in life, but come at a cost later in life.
The finding also challenges the idea that the protein produced by the HD gene is solely a toxic protein that causes brain degeneration.
“Overall, our study suggests that we should rethink the notion of the toxic protein theory,” says Nopoulos, who is also a member of the Iowa Neuroscience Institute.
“Instead, we should consider the theory of antagonistic pleiotropy—a theory that suggests that genes like HTT build a better brain early in life, but the cost of the superior brain is that it isn’t built to last and may be prone to premature or accelerating aging.
“This means that instead of knocking down the gene for therapy, drugs that slow the aging process may be more effective.”
Next steps
Nopoulos’s team is already making progress extending the research from the Kids-HD program. Nopoulos has established the Children to Adult Neurodevelopment in Gene-Expanded Huntington’s Disease (ChANGE-HD), a multi-site study that aims to recruit hundreds of participants for a total of over 1,200 assessments to validate the key findings from the Kids-HD study and to enhance future research on HD.
A primary area of focus will be understanding how an enlarged brain can later lead to degeneration. One hypothesis Nopoulos and her team will explore involves the idea that an enlarged cortex might produce excess glutamate (an important neurotransmitter), which is beneficial in early brain development, but later leads to neurotoxicity and brain degeneration.
In addition to Nopoulos, the UI team included Mohit Neema, MD, UI research scientist and first author of the study; Jordan Schultz, PharmD; Douglas Langbehn, MD, Ph.D.; Amy Conrad, Ph.D.; Eric Epping, MD, Ph.D.; and Vincent Magnotta, Ph.D.
More information: Mohit Neema et al, Mutant Huntingtin Drives Development of an Advantageous Brain Early in Life: Evidence in Support of Antagonistic Pleiotropy, Annals of Neurology (2024). DOI: 10.1002/ana.27046
Journal information: Annals of Neurology
Provided by University of Iowa
https://medicalxpress.com/news/2024-11-huntington-disease-gene-early-brain.html