
Violent blows or jolts to the head can cause traumatic brain injury (TBI), and there are currently about five million people in the U.S. living with some form of chronic impairment after suffering a TBI. Even in a mild form, TBI can lead to lifelong nerve cell deterioration associated with a wide array of neuropsychiatric conditions. Tragically, there are no medicines to protect nerve cells after injury. Behind aging and genetics, TBI is the third leading cause of Alzheimer’s disease (AD), yet the link between these two conditions is not understood.
In a new study, published online today in Cell, researchers have discovered a new way to prevent brain nerve cells from deteriorating after injury, which also revealed a potential mechanistic link between TBI and AD. Their discovery also yielded a new blood biomarker of nerve cell degeneration after injury, which is significant because there is an urgent need for mechanism-based blood biomarkers that can diagnose TBI and stage its severity.
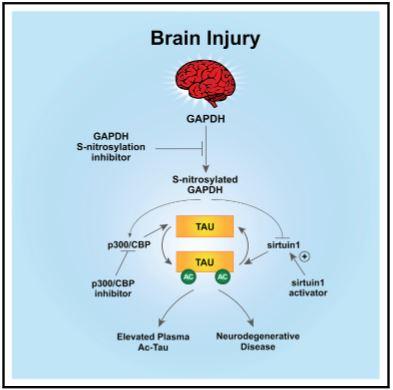
Prior to this study, it had been previously reported that a small protein in nerve cells, called tau, was modified by a chemical process called acetylation in the post-mortem brains of AD patients. But how this modification came about, as well as its role in the disease process, was not understood.
“Normally, tau functions in nerve cells to maintain the appropriate structure of the axon, which is the nerve cell extension required for nerve cells to communicate with one another,” said Andrew A. Pieper, MD, PhD, senior author on the study, Harrington Discovery Institute (HDI) Investigator and Director of the HDI Neurotherapeutics Center at University Hospitals (UH), Morley-Mather Chair in Neuropsychiatry at UH, Director of the Translational Therapeutics Core of the Cleveland Alzheimer’s Disease Research Center, and VA Geriatric Research, Educational and Clinical Care (GRECC) Investigator. “Given the relationship between AD and TBI, we wondered whether elevated acetylated-tau (ac-tau) might also occur in TBI, and if so, then whether this could provide an experimental platform to study its potential role in nerve cell deterioration.”
Dr. Pieper’s lab discovered that ac-tau increased rapidly in multiple forms of TBI in mice and rats, and persisted chronically when nerve cell degeneration was untreated. They also showed that the increased ac-tau in human AD brain was further exacerbated when the AD patient also had a prior history of TBI.
“Our research showed that after ac-tau rises, a specific structure at the junction of the nerve cell body and its axon, called the axon initial segment, breaks down,” explained Min-Kyoo Shin, PhD, co-first author of the study. “As a result, tau is no longer appropriately sequestered in axons. This leads to axonal degeneration, followed by neurologic impairment.”
The team tested therapeutic interventions after TBI at each of the three nodal points in the new signaling pathway that they identified as leading to increased nerve cell ac-tau after injury. Using known medicines or experimental drugs, they saw that all three points provided effective therapeutic opportunity.
Strikingly, they found that two FDA-approved medicines of the NSAID class (anti-inflammatory medicines commonly used as pain relievers), salsalate and diflunisal, were potently neuroprotective after TBI in mice. Relative to all other NSAIDs and distinct from their anti-inflammatory property, these two medicines inhibit the acetyltransferase enzyme in nerve cells that adds the acetyl group onto tau protein after brain injury.
Next, they examined more than seven million patient records and learned that usage of either salsalate or diflunisal was associated with decreased incidence of both AD and clinically diagnosed TBI, compared to usage of aspirin in other patients for the same time period. The protective effect was stronger in diflunisal and salsalate, which correlates with diflunisal’s superior potency in inhibiting the acetyltransferase enzyme, relative to salsalate. The NSAID aspirin was used as a comparison group because it does not inhibit the acetyltransferase.
Lastly, because the tau protein freely diffuses from the brain into the blood, the researchers examined whether ac-tau might also be elevated in the blood after TBI. In mice, they found that blood levels of ac-tau correspond tightly with brain levels, and that blood levels return to normal when mice are treated with therapeutics that lower brain ac-tau and thereby protect nerve cells. Importantly, they also found that ac-tau was significantly increased in the blood of human TBI patients.
“This work has a number of potential clinical implications,” explained Edwin Vázquez-Rosa, PhD, co-first author on the study. “First, it shows that the medicines salsalate and diflunisal provide previously unidentified neuroprotective activity by this new mechanism, and that in the course of being prescribed these medicine for traditional indications patients appear to also be relatively protected from developing neurodegenerative conditions. Accordingly, these medicines may also help protect TBI patients from developing AD. Finally, our work provides a new blood biomarker of neurodegeneration in the brain after TBI that could be harnessed to stage severity and progression of nerve cell deterioration after injury.”
Robert A. Bonomo, MD, Associate Chief of Staff at VA Northeast Ohio Healthcare System and professor at Case Western Reserve School of Medicine added, “Many of our patients suffer from TBI or AD. These important findings will have a tremendous, long-term impact on our Veteran population.”
Next steps in the research involve further investigation of the applicability of ac-tau as a biomarker in neurodegenerative disease and the potential utility of diflunisal or salsalate as neuroprotective medicines for people, as well as deeper study of the mechanisms by which ac-tau causes nerve cell deterioration.
###
The findings from the Pieper lab represent an outgrowth of collaborative efforts from investigators across the Cleveland community at UH, HDI, Case Western Reserve, VA Northeast Ohio Healthcare System, Cleveland Clinic and The MetroHealth System.
This study was also supported by the Brockman Foundation; the AHA/Allen Initiative in Brain Health and Cognitive Impairment; The Neuropathology Core of Northwestern University; Translational Therapeutics Core of the Cleveland Alzheimer’s Disease Research Center; and the Departments of Neurology and Neurosurgery of McGovern Medical School of The University of Texas Health Science Center at Houston.
Shin, M, Vázquez-Rosa, E., et al. “Reducing acetylated-tau is neuroprotective in brain injury.” Cell. DOI: 10.1016/j.cell.2021.03.032.
