by SUKANYA CHARUCHANDRA
It wasn’t until the latter half of the 13th century that human dissections became acceptable in Italy. Previously, both the Roman Empire and Islamic law had prevented the dissection of humans and its depiction. While the Greek surgeon Galen’s anatomical drawings from the second century had been preserved and studied until the Renaissance, they were largely based on dissections of animals, such as apes.
In the mid-16th century, however, famed Flemish anatomist Andreas Vesalius dissected the bodies of executed criminals—not an uncommon practice in that period—while studying in Paris. He realized that Galen had been “misled” by apes, whose anatomy was not exactly like that of humans.
“The challenge of anatomy is rendering the 3-D experience of opening bodies onto a 2-D page,” writes Hannah Marcus, a science historian at Harvard University, in an email to The Scientist. Lack of refrigeration also presented a challenge. In overcoming those hurdles to produce the first realistic depictions of internal human biology, Vesalius’s De Humani Corporis Fabrica, published in Basel, Switzerland, in 1543, galvanized the study of anatomy.
Meanwhile, Spanish-born Juan Valverde de Amusco was learning anatomy under the guidance of Roman surgeon Realdo Colombo, and possibly of Vesalius himself, at the University of Padua in Italy. Valverde observed and participated in many dissections under Colombo’s guidance, and pored over old books on the subject. He later moved to Rome and was welcomed into the home of Spanish Cardinal Juan Álvarez de Toledo.
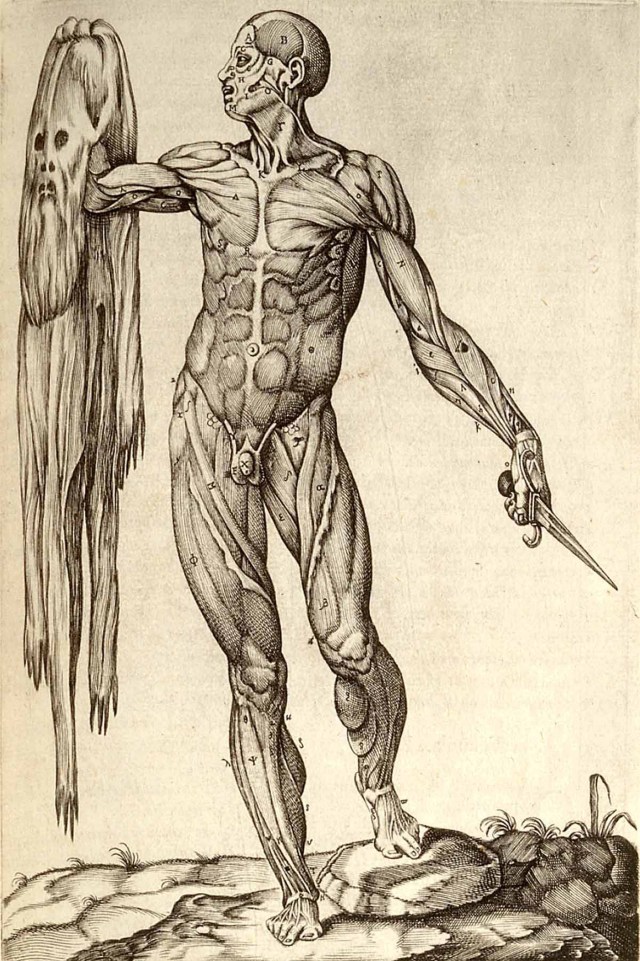
In 1555, Valverde served as a doctor at the foremost contemporaneous Roman hospital, Santo Spirito, where many luminaries of anatomy worked during that period, including Bartolomeo Eustachi, under whom Valverde studied for a time. The following year, Valverde crafted the Spanish-language anatomical text Historia de la Composicion del Cuerpo Humano, or Account of the Composition of the Human Body. In seven parts, the book covered topics such as “bone and cartilage,” “ligaments and bandaging,” and “instruments of sensation and external motion.” Largely copied from the 1543 and 1555 editions of Vesalius’s tome, it included 15 new illustrations in four copper plates. Valverde’s book also included more than 60 corrections to Vesalius’s text, which enhanced the contemporary understanding of the intracranial passage of carotid arteries, the extraocular muscles, the stapes bone of the middle ear, and how blood moves through the septum. Historians attribute the few original illustrations to Spanish-born Gaspar Becerra.
“Vesalius was angry about Amusco’s work and accused him of plagiarism,” Marcus writes. In 1564, Vesalius wrote in his book Anatomicarum Gabrielis Fallopii Observationum Examen that “Valverde who never put his hand to a dissection and is ignorant of medicine as well as of the primary disciplines, undertook to expound our art in the Spanish language only for the sake of shameful profit.” Valverde conceded his borrowing, explaining that Vesalius’s drawings were so thorough that “it would look like envy or malignity not to take advantage of them.”
Valverde simplified Vesalius’s Latin text considerably, however, as he considered it difficult to understand. His more concise (and thus cheaper) text had more than a dozen editions published in Italian, Latin, Dutch, and Greek, in addition to Spanish, and facilitated the spread of scientific ideas and Vesalius’s modern anatomy throughout Europe and the Spanish Americas.
https://www.the-scientist.com/foundations/homo-sapiens-exposed–1556-64679